

Daniel Sucerquia
How a Stretching Force Differently Destabilizes Chemical Bonds on a Protein Backbone
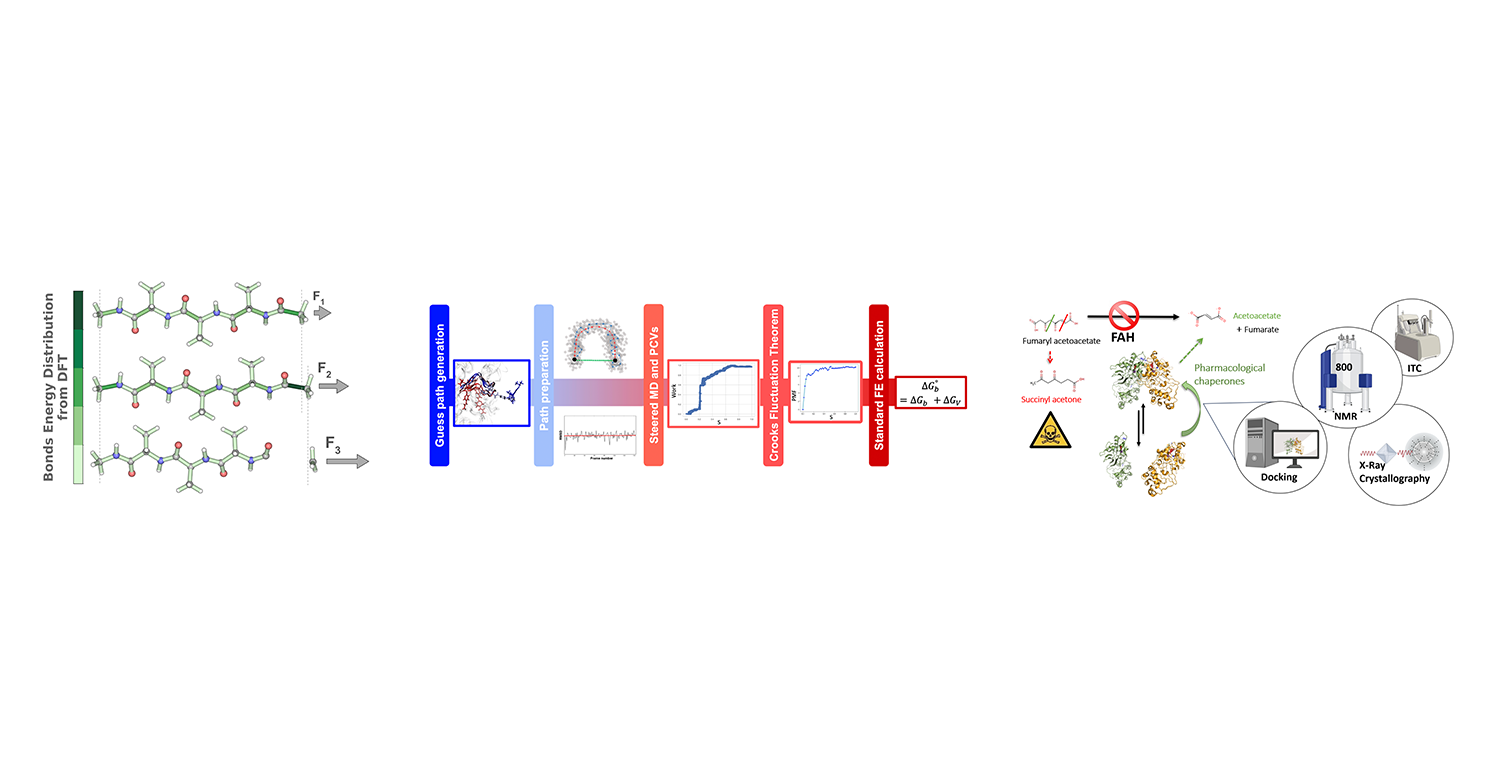
When subjecting a protein chain to extreme pulling forces, bonds in the stretched backbone ultimately break. As a most simple assumption, a protein backbone can be considered as a serie of springs each of which carries the same force. However, proteins are more complex than that and force will distribute across the various degrees of freedoms in the peptide, largely depending on the chemical environment. We here study the changes of energy stored in the degrees of freedom of molecules at quantum level of accuracy. We so far have tested this method in chains of 3 amino acids. We observe a linear increase in energies per degree of freedom including bonds, angles, and dihedrals, during stretching, and proline to show an energy distribution distinct from the other amino acids due to the ring structure. Data collected with this method applied to a large set of small peptides will aid to predict the energy distribution in larger systems using Machine Learning.
Biography
I am originally from Colombia, where I studied my bachelor and master in physics at the University of Antioquia. During my bachelor and master, I studied stability of silver clusters using ab-initio and enhanced sampling approximations. That research was done with the advice of Professor Olga L. López and Dr. Pilar Cossio. Since a year ago, I moved to Heidelberg, Germany, to do my PhD at the Heidelberg institute for Theoretical Studies at the Molecular Biomechanics group (MBM), led by professor Frauke Gräter.
Social Media
Personal twitter: @sucer6
Twitter of the institute: @HITStudies
Twitter of the project: @simplaix


Eleonora Serra
Bi-directional path-based nonequilibrium simulations for binding free energy estimation
This work presents a novel pipeline that combines path collective variables (PCVs) obtained through enhanced sampling and machine learning with two-directional Steered Molecular Dynamics (SMD) to estimate protein-ligand binding free energy. We validated our workflow on a host-guest system and one protein-ligand complexes, achieving good accuracy and precision between our computed results and experimental data.
Biography
I am a PhD student at IIT (Fondazione Istituto Italiano di Tecnologia, 16163 Genova, Italy) in the Computational and Chemical Biology group headed by Andrea Cavalli, and I am working in collaboration with thecomputational group of the Centre Européen de Calcul Atomique et Moléculaire(CECAM), Lausanne. Our interest is focused on finding new computational strategies for computing binding Free Energy. I took my bachelor’s degree in chemistry from the “Università degli studi di Genova” (Curriculum Chimica e Scienze Chimiche), and I have a Chemistry and Bioinformatic Master Degree : Chemistry and In Silico DrugDesign for Macromolecules. In particular, I followed a Double-degree international program having 2 different specialties: Degree of “Laurea magistrale in Scienze Chimiche” from Università degli studi di Milano and Degree of “Master in Chemistry, specialty Chemoinformatics” from University of Paris. Moreover, I had the opportunity to do my master’s internship at IKTOS (Paris), which is a company mostly working on developing new AI technologies for In-SilicoDrug Design


Riccardo Scarin
Development of pharmacological chaperones for the treatment of tyrosinemia type 1
Tyrosinemia type 1 is a genetic disease characterized by several mutations of FAH gene that leads to the unfolding of the enzyme fumaryl-acetoacetate hydrolase (FAH) and a consecutive block of tyrosine catabolism. The aim of this project is to develop a treatment based on a pharmacological chaperone that restore the functionality of the enzyme by stabilizing the native state of FAH. This is accomplished by using biophysical NMR techniques, computational techniques and crystallography. So far, through our research, we could design lead compounds that bind in the lower micromolar and nanomolar range. These compounds have shown, in biophysical assays, to stabilize one of the mutated forms of FAH involved in the pathology.
Biography
I am Riccardo Scarin, a 26-year-old Italian Ph.D. student in the Marie-Curie ITN GLYTUNES program. My interest has always revolved around comprehending molecular interactions, which is my primary motivation in research. Currently, under the guidance of Dr. Oscar Millet at ATLAS Molecular Pharma SL and CICBiogune (Spain), I am developing molecular chaperones for the treatment of rare diseases. My tasks involve protein expression and purification, followed by the characterization of ligand binding using 2D-NMR, X-ray crystallography, and computational approaches. I hold a Bachelor's degree in Pharmaceutical Science and a Master's degree in Pharmaceutical Biotechnology from the University of Padua (UNIPD). During my career I did Internships at:
• Laboratory of Prof. Stefano Moro (UNIPD), focusing on cheminformatics;
• Edmund Mach Foundation (Trento), in Metabolomics section (Dr. Stefan Martens Lab);
• University of Vienna (Prof. Christoph Rademacher), in fragment screening by NMR;
• Max Planck Institute– Glycouniverse GmbH (Potsdam), in Organic Synthesis.
My hobbies include playing trumpet, bamboo flutes, guitar and practicing Yoga and sports.